.png)
“Borderline products” are products having combined characteristics of medicines along with foods, medical devices or cosmetics. The following criteria shall be taken into consideration when evaluating borderline products for registration
• The intended use of the product (or its primary function) and its mode of action
• The therapeutic claims made by the manufacturer regarding the product in terms of its ability to treat or prevent diseases
• Pharmacologically active substance(s), if any, used in the product.
• The concentration of the active substances
• The level of efficacy of the active substance of the product
• The ingredients used and the concentrations at which they are used.
According to the National Medicines Regulatory Authority Act No. 05 of 2015 the National Medicines Regulatory Authority shall be responsible for the regulation and control of registration, licensing, manufacturing, importation and all other aspects pertaining to borderline products; The Borderline Products Evaluation Committee (BPEC) will carry out the technical evaluation of products submitted for registration and assist the Authority to regulate and control all aspects pertaining to borderline products.
The National Medicines Regulatory Authority (NMRA) is responsible to build a healthier nation by ensuring the availability of quality and safe borderline products in Sri Lanka. As such all products which may come under Borderline Product category should be registered with the Authority.
All foreign borderline products manufactures should submit application for registration through a Marketing Authorization Holder (local agent) in Sri Lanka who shall be responsible for the registration, licensing, importation, sale and distribution, handing of quality failures and all aspects pertaining to the particular borderline product in Sri Lanka.
Borderline products have been classified to three schedules by the BPR division in the registration process depending on the level of risk and to make sure the awareness of the general public regarding the level of risk of such products.

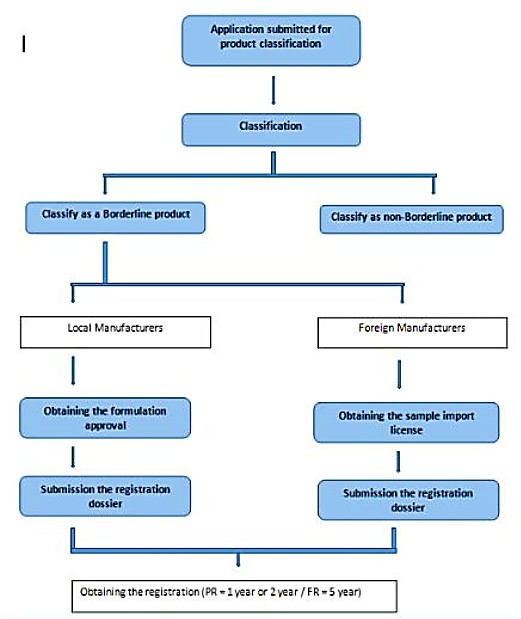
How To Register Your Product
The Product Registration Process Is In Two Steps:
I. Preliminary Evaluation (Product Classification) –Valid period (only 1 year)
II. Product Registration- Valid period(1 year, 2 year or 5 years)
Local Agent Profile Creation / Local Manufacturing Profile Creation and obtaining a Preliminary Evaluation Report(Product Classification Report) are mandatory requirements to initiate the product registration process and application submission should be done throughe NMRA facility. Before the preliminary evaluation, local agent / local manufacturing profile creation is a main requirement. (via eNMRA platform). Except Preliminary Evaluation (Product Classification) and profile creation all the other applications should be submitted manually.
Currently this process (Local Agent Profile Creation / Local Manufacturing Profile Creation)has been hold temporally due to online system failure and all the submissions should be done manually.
Steps in Borderline Product Registration:
1. Preliminary evaluation (Product Classification)
2. Formulation approval (applicable only for local manufacturers) or sample import license (applicable only for foreign manufacturers)
3. Product Registration (Local /Foreign)
1.Preliminary Evaluation (Product Classification)
1.1 Required documents for “Preliminary Evaluation(Product Classification)” under Borderline Product Category
1. Covering letter including contact details of the local agent (e-mail address, contact numbers, address/s)
2. A valid copy of Letter of Authorization from the manufacturer which addressed to Director General / CEO ,NMRA (If more than one local agent present for a manufacturer, the list of local agents appointed by the manufacturer and approved product list - for local agents only)
3. Product master formula and Batch manufacturing formula *
4. Valid copy of Free Sale Certificate(FSC) or Certificate of Pharmaceutical product (COPP) (Not applicable for locally manufactured products).
The FSC/COPP must comply with the following requirements:
5. Declaration form
6. Copy of acceptable Certificate of Analysis for Finished Product
7. Legible specimen colored label,Packaging materials (art work acceptable)
8. Legible product information leaflet/ pack insert
9. Promotional materials (If applicable only)
* All applicants who submit applications under Borderline products should submit the composition of unit dose (Product master formula) along with the batch manufacturing formula. If overages are added, the quantities and percentages must be specified.
Vitamins and minerals should be provided according to the “Guideline on Product Categorization Reference details of Vitamins & Elements” publish in the NMRA website under borderline products category. (Units and form of vitamin & minerals)
Examples:
• Elemental –Sodium (Sodium should be given as “Na” but not as “NaCl”)
• Vitamins-Vitamin A (Vitamin A should be given as “vitamin A” but not as “retinol’)
• Units- Vitamin D in μg/day, Zn inmg/day)
Refer: The Borderline Product Guideline - “Guidance Document on definitions and information for applications submitted under Borderline Product category”
For further Information click here
All non compliant applications will be rejected with effect from 13th of July 2020.
After the classification process,Classification Report will be issued. Validity period of Preliminary Evaluation Report (Classification Report) is one year and product registration process should be initiated within the validity period of the Classification Report.
2. Formulation Approval and Sample Import License
2.1 Required documents for obtaining the formulation approval (Only for Local Manufacturers)
1. Preliminary evaluation report(Classification Report)
2. Covering letter including contact details of the local agent (e-mail address, contact numbers, address/s)
3. Formulation (Master formula &Batch manufacturing formula)
4. Legible product information leaflet/pack insert
5. Legible specimen colored label,Packaging materials (art work acceptable)
2.2 Required documents for obtaining the Sample Import License (for imported products)
1. Preliminary Evaluation Report(Classification Report) issued by NMRA
2. Application for a License to import a limited quantity of any borderline product(s) for testing, examination,distribution, sample analysis or clinical trial
3. Copy of Business Registration Certificate.
4. Copy of Letter of Authorization from the manufacturer which addressed to Director General / CEO, NMRA (If more than one local agent present for a manufacturer, the list of local agents appointed by the manufacturer and approved product list).
5. Catalogue (If applicable only)
6. Valid Copy of Free Sale Certificate(FSC) /Copy of Certificate of Pharmaceutical Product (COPP), along with the requirements stated in point 4 of section 1.1.
7. Specimen colored label and packaging materials
3.0 Required documents for product Registration
3.1 Local Manufactured products
1. Acknowledgment form – 02 copies
2. Declaration letter
3. Borderline Classification Report issued by NMRA
4. Formulation approval letter – for new registration
5. Previous Registration certificate –for re registration
6. Maximum retail price
7. Business registration certificate
8. Wholesale License (If available).
9. GMP report or GMP certificate for manufacturing site
10. ISO certificate (If GMP certificate is not available)
11. Valid manufacturing license with the product list.
12. Registration certificates of other countries (If available)
13. Finish product test specifications
14. Acceptable Certificate of analysis for finished Product (Original)
15. Acceptable Certificate of Analysis of Active Pharmaceutical Ingredient (API)
16. Valid API GMP certificate(s) along with the approved API list from the API manufacturer(s)
17. Acceptable Certificate of Analysis of excipients (If applicable)
18. Product master formula and batch manufacturing formula must be submitted, including the composition of the unit dose, along with the function and specification of each ingredient. If overages are added, the quantities and percentages must be specified.
19. Manufacturing process with process flow diagram clearly
20. Manufacturing process validation report for three consecutive commercial batches (If applicable only)
21. Analytical validation report (if applicable only)
22. Stability data for three commercial batches (Real-Time and Accelerated) – Original documents or Shelf life declaration form as per Product Schedule (if applicable)
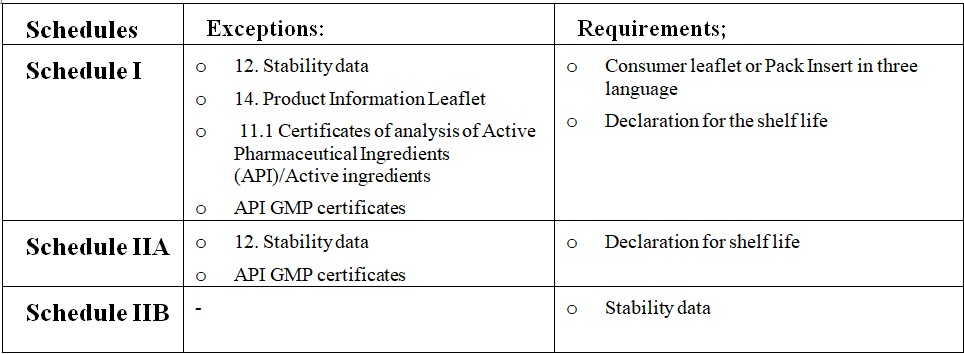
23. Specifications & Certificates of Analysis of Packaging materials
24. Product Information Leaflet/ pack insert
25. Efficacy data (If applicable only)
26. Packaging materials- Specimen colored label & six side displayed secondary package (If applicable only)
27. Promotional Material (If applicable only)
28. Two samples
29. Post marketing data (If applicable only)
30. Published Clinical trial data for finish product (If applicable only)
31. Catalogue (If applicable only)
3.2 For Imported Products
1. Acknowledgment form – 02 copies
2. Declaration letter
3. Letter of Authorization from the manufacturer which addressed to Director General / CEO, NMRA (If more than one local agent present for a manufacturer, the list of local agents appointed by the manufacturer and approved product list)
4. Borderline Classification Report issued by NMRA
5. Sample Import License – for new registration
6. Previous Registration certificate –for re registration
7. Maximum retail price
8. Business registration certificate
9. Wholesale License (If available)
10. Valid original of Free Sale Certificate (FSC) /Certificate of Pharmaceutical product (COPP), along with the requirements stated in point 4 of section 1.1.
11. GMP report or GMP certificate for manufacturing site
12. ISO certificate (If GMP certificate is not available)
13. Manufacturing License with the product list
14. Registration certificates of other countries (If available only)
15. Finish product test specifications
16. Acceptable Certificate of analysis for finished Product (Original)
17. Acceptable Certificate of Analysis of API
18. Valid API GMP certificate(s) along with the approved API list from the API manufacturer(s)
19. Acceptable Certificate of Analysis of excipients
20. Product master formula and batch manufacturing formula must be submitted, including the composition of the unit dose, along with the function and specification of each ingredient. If overages are added, the quantities and percentages must be specified.
21. Manufacturing process with process flow diagram clearly
22. Manufacturing process validation report for three consecutive commercial batches (If applicable only)
23. Analytical validation report (if applicable only)
24. Stability data for three commercial batches (Real-Time and Accelerated) – Original documents or Shelf life declaration form as per Product Schedule (if applicable)
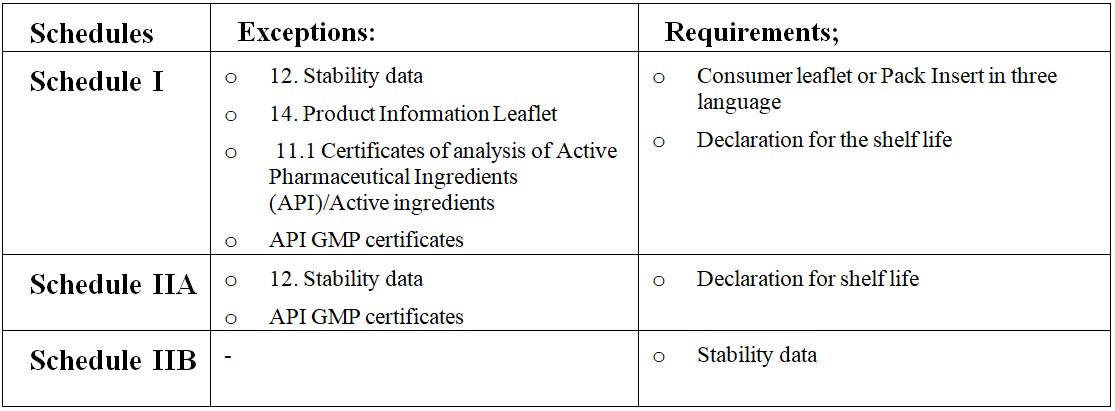
25. Specifications & Certificates of Analysis of Packaging materials
26. Product Information Leaflet/ pack insert
27. Efficacy data (If applicable only)
28. Packaging materials- Specimen colored label & six side displayed secondary package (If applicable only)
29. Promotional Material (If applicable only)
30. Two samples
31. Post marketing data (If applicable only)
32. Published Clinical trial data for finish product (If applicable only)
33. Catalogue (If applicable only)
Please refer to the guidelines published for borderline products, as well as the web notices and applicable forms.
Preliminary Evaluation (Product Classification)
Required documents for “Preliminary Evaluation (Product Classification)” under Borderline Product Category
1. Covering letter including contact details of the local agent (e-mail address, contact numbers, address/s)
2. A valid copy of Letter of Authorization from the manufacturer which addressed to Director General / CEO , NMRA (If more than one local agent present for a manufacturer,the list of local agents appointed by the manufacturer and approved product list - for local agents only)
3. Product master formula and Batch manufacturing formula *
4. Valid copy of Free Sale Certificate (FSC) or Certificate of Pharmaceutical product (COPP) (Not applicable for locally manufactured products).
The FSC/COPP must comply with the following requirements:
5. Declaration form
6. Copy of acceptable Certificate of Analysis for Finished Product
7. Legible specimen colored label, Packaging materials (art work acceptable)
8. Legible product information leaflet/ pack insert
9. Promotional materials (If applicable only)
* All applicants who submit applications under Borderline products should submit the composition of unit dose (Product master formula) along with the batch manufacturing formula.If overages are added, the quantities and percentages must be specified.
Vitamins and minerals should be provided according to the “Guideline on Product Categorization Reference details of Vitamins & Elements” publish in the NMRA website under borderline products category. (Units and form of vitamin & minerals)
Examples:
• Elemental –Sodium(Sodium should be given as “Na” but not as “NaCl”)
• Vitamins-Vitamin A(Vitamin A should be given as “vitamin A” but not as “retinol’)
• Units- Vitamin D in μg/day, Zn in mg/day)
Refer: The Borderline Product Guideline - “Guidance Document on definitions and information for applications submitted under Borderline Product category”
For further Information click here
All non compliant applications will be rejected with effect from 13th of July 2020.
After the classification process, Classification Report will be issued. Validity periodof Preliminary Evaluation Report (Classification Report) is one year andproduct registration process should be initiated within the validity period ofthe Classification Report.
Approval of Foreign Manufacturing Facilities
Introduction & Scope:
The process applies for overseas manufacturing sites who intended to get marketing authorization for their products in Sri Lanka. The procedure includes a desk review of essential documents including the site master file. A GMP inspection of the site may be necessary before approval of the site.
A foreign manufacturer who wants to register products in Sri Lanka needs to appoint a Sri Lankan business entity as its local authorized agent. The local agent will furnish applications on behalf of its principal company, the foreign manufacturer. A separate application needs to be submitted to each manufacturing site from which the manufacturer plans to export products to Sri Lanka.
Application has to be submitted online via NMRA’s web portal eNMRA which requires uploading of essential information and documents to enable a desk review. If all documents indicate fulfilment of NMRA requirements, the next step would be to carry out a GMP inspection of the facility. The NMRA Act, No. 5 of 2015 (section 51) & regulations 2 to 10 of part I of medicines regulations pronounce the legal requirements. The NMRA would exempt onsite GMP inspection if the particular site had been inspected by a stringent NRA or WHO, in terms of regulation 24 of the medicines regulations. After these procedures, an approved foreign manufacturer can submit an application for marketing authorization of its products through the local agent.
Flow Chart- Foreign Manufacturing Site Approval
The process applies for local manufacturing sites who intended to get marketing authorization for their products in Sri Lanka. The procedure includes a desk review of essential documents including the site master file and several GMP Inspections.
A local manufacturer who wants to register products in Sri Lanka needs to furnish applications regarding his manufacturing facilities. A separate application needs to be submitted to each manufacturing site.
Application has to be submitted online via NMRA’s web portal eNMRA which requires uploading of essential information and documents to enable a desk review and GMP Inspections.
The NMRA Act, No. 5 of 2015 (section 51) & regulations 26 to 30 of part II of medicines regulations pronounce the legal requirements.
After these procedures, an approved Local manufacturer can submit application for formulation approvals.
After obtaining the formulation approval and GMP approval for commercial manufacturing of the facility, local manufacturer can apply for the product registration. After that, local manufacturer can apply for the manufacturing license.
Site Master Files are submissions to NMRA used to provide confidential, detailed information about facilities, processes, or articles used in the manufacturing, processing, packaging, and storing of Medicines, Medical Devices, Borderline products and cosmetics.
Guidance for Site master Files
References:
Good manufacturing practice (GMP) is that part of quality assurance which ensures that products are consistently produced and controlled to the quality standards appropriate to their intended use and as required by the marketing authorization.
The National Medicines Regulatory Authority Act No. 5 of 2015 interprets GMP guidelines as ‘good manufacturing guidelines issued by the World Health Organization’. As such, the National Medicines Regulatory Authority (NMRA) adopts the WHO GMP guidelines along with its subsequent updates for the purpose of regulation of pharmaceutical manufacture. The manufacturers are expected to adhere to the Good Manufacturing Practices throughout their activities.
Pharmaceutical manufacturers who intend to apply for marketing authorization in Sri Lanka may be subjected to one or several GMP inspections prior to their site approvals.
GMP Guidelines available on WHO website:
WHO good manufacturing practices for pharmaceutical products: main principles:
https://www.who.int/medicines/areas/quality_safety/quality_assurance/TRS986annex2.pdf?ua=1
WHO good manufacturing practices for sterile pharmaceutical products:
WHO good manufacturing practices for biological products:
https://www.who.int/publications/m/item/annex-2-WHO-gmp-for-biological-products
Guideline on Approval of an Overseas Manufacturing Plant of Medicines
Guideline on Preparation of Site Master Files
Guideline for preparation and conduct of the GMP inspection
Healthcare professional such as doctors, dentists, pharmacists and nurses are encouraged to report suspected adverse events encountered in their day to day practice.
Using the ADR google form given by the following link here, you can report Adverse Events.
Adverse Drug events reporting
Address
Director General / CEO
National Medicines Regulatory Authority,
No: 120,
Norris Canal Road,
Colombo 10.
Telephone
+94 112 698 896, +94 112 698 897
Fax
+94 112 689 704
A copy of the form shall be forwarded to Adverse Drug Reaction Monitoring Unit of the Department of Pharmacology, Faculty of Medicine, University of Colombo, Kynsey Road, Colombo 08. (TP/ Fax: +94 112 697 483).
When a new chemical entity (NCE) is first marketed, it would have been tested only in a limited number of patients/volunteers. Rare adverse drug reactions/events could be identified only after the drug is marketed and used by a much larger population. Safety information in use in special groups such as children, elderly, pregnant women etc. are not often available at the time of first marketing of a new drug.

The Advertising Evaluation Sub Committee of the NMRA regulates advertisements - printed and electronic material - related to medicines, devices, borderline products to ensure such advertisements are ethical, accurate and appropriate.
Working definition & general rules
Advertisement includes any representation by any means whatsoever, for the purpose of promoting directly or indirectly the manufacture, sale or disposal of any medicine, medical device, borderline product.
Our Scope & Responsibility
Procedure for applying approval for advertisements
All the applicants who which to obtain approval for the advertisement on medicines, medical device and borderline products should be followed below procedure.
1. Submit the duly filled application (Please mention email address correctly for sending evaluation sheet) along with copy of valid registration certificate, story board of the advertisement to the accepting counter on every Friday (From 9.00 am to 3.30 pm)for obtaining the payment note for processing fee (1000USD).
2. Payment Receipt issued by account division should be submitted to NMRA after the payment along with the application with following documents to accepting pharmacist (Every Friday from 9.00 am to 3.30 pm). Application should be submitted in a file. Accepting pharmacist accepts the application and assign a reference number.
Documents Needed to be enclosed with the application :-
3. All the applications will be tabled to the committee (Once a month) and evaluation reports will be emailed to the applicants.
4. Approved letters can be collected from the issuing counter.in the advertisements
Applicants should consider the following when designing an advertisement:
F-BPR-001 Revised Check List for Evaluation of New Borderline Product Application
F-BPR-02 WOR Application - Borderline Products
F-BPR-03 WOR Assessment form - Borderline Products
F-BPR 008 Application for a Variation-Borderline Products
F-BPR-009 Checklist for accepting Classification of Borderline Products
F-BPR-010 Checklist for accepting Registration Applications
F-BPR-011 Acknowledgment form of Borderline Products
F-BPR-013 Declaration form for Shelf Life
Application for a License to Import a Limited Quantity of any Borderline Product
Declaration of the Authorized person
Archived guidelines
Current issues related to the products coming under the category of borderline products;
A. Classification application submission and customs release for the products which may come under the borderline product category
As per the NMRA Act, since 2016, the Borderline Product Regulatory Division has being continuously involved in the product registration. Majority of importers have brought such products to the country without obtaining the product registration from NMRA and recently Sri Lanka Customs introduced an online platform called as ASYCUDA SYSTEM to release the products linking with other relevant government regulatory bodies.
Once introducing this ASYCUDA SYSTEM, many importers were unable to release their products through the system due to unavailability of product registration. As a result of, it creates a gap between a regulatory perspectives and continuation of market field causing an accumulation of goods at Sri Lanka Customs.
For this matter following solutions were proposed:
1. Products release through the ASYCUDA SYSTEM only;
Products that fall under the borderline category will be released only through the ASYCUDA system, and registration is a mandatory requirement. However, a deadline was proposed for the classification applications submission: April 30, 2025 to release the product through the ASYCUDA system without the product registration (Classification application submission is mandatory ) and the consignments could be released until 31, October 2025
2. Normal registration approval process will be effective for the applications submitted from May 1, 2025, and product release will be conducted through the ASYCUDA system only for the registered products.
3. Anyhow during the registration process (including the classification step) if any product is rejected, the consignment clearance will be discontinued (for 1 & 2).
4. For consideration of custom clearance of a product, it is mandatory to upload the acknowledgment form/s of application submission (Classification, Sample Import license and Registration Dossier) 3. Offence against the misleading/incorrect information found in a product:
5. Offence against the misleading/incorrect products;
Any misleading/incorrect information (related to a product) in an advertisement, on the label, PIL, packaging materials or in any other manner will be punishable offence under the requirements of the NMRA Act, No. 5 of 2015.
B. Publication of new guideline related in borderline products
A guideline will be published in due course for the following list of products, including TUL(Tolerable Upper Levels), Minimum, and Maximum levels. Once the guideline is published, adherence to the guideline will be required for the continuation of product registration
List of ingredients :Fluoride, Probiotics, Chondroitin, D-Mannose, Alpha-lipoic acid (ALA),Choline, Glucosamine, Co-enzyme Q10, Inositol, Omega 3 (EPA +DHA), Omega 6 (gamma Linoleic acid), Taurine, N-Acetyl cysteine, Methyl Sulphonyl Methane (MSM), N-Acetyl L-tyrosine, Vitamin K2, Phosphatidycholine (Lecithin), Carnitine and Boron
C. Export only products;
Export only products will not be considered for product registration under the category of borderline products.
Exceptions: product registration in reference NRAs
D. Model label for registered borderline products
A sticker or label on the commercial pack will be mandatory for all registered borderline products, with effective from August 31, 2025.
Model label:

1. Classification Additional
The valid period of Classification Report is one year and the valid period is counted from the approval date of the Classification Report ("Classified as a Borderline Product").
Only two additional data submissions are allowed for one Classification Application.
If a Classification Report is issued as a "awaiting additional data" for a new application;
2. Sample Import Licence/ Formulation Approval
Sample Import Licence/ Formulation Approval requesting application to be submitted to NMRA within one year period from the date of approval of the Classification Report classified as a Borderline Product.
The valid period of Sample Import licence/ Formulation approval application is one year
3. Registration Dossier Submission.
Registration dossier to be submitted within the valid period of the Sample Import Licence/Formulation Approval.
Notes:
If any applicant is failed to submit any of the above applications (1,2 & 3) within the valid period is subjected to reject the consignment clearance approval.
National Medicines Regulatory Authority,
State Engineering Corporation Building (2nd Floor),
No. 130,
W.A.D Ramanayaka Mawatha,
Colombo 02,
Sri Lanka