.png)
According to the National Medicines Regulatory Authority Act No. 05 of 2015 the National Medicines Regulatory Authority shall be responsible for the regulation and control of registration, licensing, manufacture, importation and all other aspects pertaining to medical devices in a manner compatible with the National Medicines Policy;
As defined in the Act a “medical device” means any instrument, apparatus, appliance, software, material or any other article, whether used single or in combination, including the software necessary for its proper application intended by the manufacturer used in or on human beings for the purpose of:
and which does not achieve its intended action in or on the human body by pharmacological, immunological or metabolic means but which may be assisted in its function by such means;
A medical device does not include an Ayurveda device or a Homeopathy device;
The National Medicines Regulatory Authority (NMRA) is responsible to build a healthier nation by ensuring that medical devices available for supply in Sri Lanka are of acceptable quality, safety and fit for their intended purpose. As such all Medical devices which are categorized as in above definition and should be registered with the Authority and license to be obtained for manufacturing, importation, re-packaging, sale, distribution and offered for sale in Sri Lanka.All foreign medical device manufactures should submit application for registration through a Marketing Authorization Holder (local agent) in Sri Lanka who shall be responsible for the registration, licensing, importation, sale and distribution, handing of quality failures and all aspects pertaining to the particular medical device in Sri Lanka.
The Medical Device Evaluation Committee (MDEC) formed under NMRA Act carries out technical evaluation of the medical devices forwarded for registration by considering the quality, safety, effectiveness, need and cost of such devices. The MDEC consist of experts drawn from various specialties in medical and pharmaceutical fields who meets monthly to decide on applications submitted for marketing authorization of medicines and to make policy decisions relevant to marketing authorization of medicines.
Responsibility of the Marketing Authorization Holder
This is to inform you that Sri Lanka Orthopedic Association has submit a new guideline for classification of orthopedic implants and instruments for the purpose of submission of applications for sample import license and registration to NMRA (Annex 1). As such you should follow below mention procedure for submission of applications for orthopedic implants and instruments.
Step 1 - Categorized the implants or instruments to main categories
(i) Orthopedic implants or instruments
(ii) Oral Maxillofacial (OMF) implants or instruments
(iii) Neuro implants or instruments
Step 2 - Classification of main group
For orthopedic products separate dossiers to be submitted according to the classification for main six (6) groups.
Step 3 - Classification of sub group
Sub groups intended to register under each main group should be clearly categorized by the manufacturer based on the Free Sale Certificate.
Eg: Main group - Adult plates and screws.
Sub groups - Large fragment locking plates and screws, Mini fragment locking plates and screws, small fragment locking plates and screws.
The NMRA will coordinate with the applicants those who has already submitted registration dossiers for re-arrangement according to the above guideline requirements.
A medical device consisting a collection of devices and has a common intended purpose is registered as a group.
Electro surgical unit with standard accessories (electrodes, electrode holders, leads, Plates, plug adopter)
Anesthesia machine with standard accessories
Nebulizer system
General Documents/ Requirements
Fulfilled Schedule I, Form A & Form B
Following documents should be submitted in addition to the basic documents where necessary / if available
Final product inspection report (for electro medical equipment and machines) and finished product test report for other products Submit relevant report issued by the manufacturer or third-party laboratory for batch release of the product
Test reports for below mentioned items
Material test report for sutures, medical instruments such as forceps, scissors etc.
Certification for quality management system according to ISO 13485 from authorized notified body in order to access the design, development, manufacturing as well as for post marketing monitoring of safety and performance of the manufacturer
CE accreditation from authorized notified body in order to prove the quality assurance system of the product and EC design examination certificate (if applicable)
Stability data for entire shelf life of the finished products should be provided (if applicable)
In addition, following requirements should be fulfilled for Absorbable sutures with the application
Details of the raw material sources, purchasing details should be provided
Where necessary certificate of approval from relevant authorities should be provided
For radiation emitting devices approval obtained from Atomic energy Authority of Sri Lanka
Certification from the relevant health authority of the country of manufacturer that the product is free from BSE (Bovine Spongiform Encephalopathy) should be obtained for animal derived products
Surgical Catgut
Biological evaluation/biocompatibility test report of medical device as per ISO standards (if applicable)
Risk management analysis as per ISO standard (if applicable)
Recently issued validation report for sterilization process for two commercial batches (if applicable)
Name of product [Approved name and brand name (if any)]
Name and address of the actual manufacturer
Whether the product is sterile and mode of sterilization
Storage conditions specifying the temperature
Manufacturing date, Expiry date and Lot no/ batch no. (if applicable)
Patient information Leaflet
Product information leaflet for the products which are individually handled by the patient in the household should be in both Sinhala and Tamil languages
Eg : Glucometers, Hearing aids, spacer device etc.
The Provisional Registration for a period of one year (or two) will be issued for first time registration and is specified in the certificate
The Full Registration of a product is valid for a period of five years and is specified in the certificate
When additional data are requested, the applicant will have to furnish additional information requested by the authority within 3 months to facilitate further evaluation
If the product is rejected, the market authorization holder will be able to appeal for registration
Application for renewal should be made before six months from the date of expiry of registration
A grace period will extend until the decision is given to the application for renewal
If the requirements for registration are not satisfactory the application will be rejected completely
Any change in product name, product specifications, packaging, indications, contents of product label, package insert, or product literature, or any relevant particulars of the registered product should not be made without the prior approval of the authority.
The registration of the product maybe cancelled if changes are made without the prior approval of the authority.
Any change of product which affects quality, safety & efficacy of the product should require a new application for registration.
This section explains the process for approval of manufacturers who would be eligible to apply for registration of their products in Sri Lanka.
Information is also given to provide guidance on the type of applications and how to apply for the approval of relevant manufacturing facilities.
Introduction & Scope:
The process applies for overseas manufacturing sites who intended to get marketing authorization for their products in Sri Lanka. The procedure includes a desk review of essential documents including the site master file. A GMP inspection of the site may be necessary before approval of the site.
A foreign manufacturer who wants to register products in Sri Lanka needs to appoint a Sri Lankan business entity as its local authorized agent. The local agent will furnish applications on behalf of its principal company, the foreign manufacturer. A separate application needs to be submitted to each manufacturing site from which the manufacturer plans to export products to Sri Lanka.
Application has to be submitted online via NMRA’s web portal eNMRA which requires uploading of essential information and documents to enable a desk review. If all documents indicate fulfilment of NMRA requirements, the next step would be to carry out a GMP inspection of the facility. The NMRA Act, No. 5 of 2015 (section 51) & regulations 2 to 10 of part I of medicines regulations pronounce the legal requirements. The NMRA would exempt onsite GMP inspection if the particular site had been inspected by a stringent NRA or WHO, in terms of regulation 24 of the medicines regulations. After these procedures, an approved foreign manufacturer can submit an application for marketing authorization of its products through the local agent.
Flow Chart- Foreign Manufacturing Site Approval
The process applies for local manufacturing sites who intended to get marketing authorization for their products in Sri Lanka. The procedure includes a desk review of essential documents including the site master file and several GMP Inspections.
A local manufacturer who wants to register products in Sri Lanka needs to furnish applications regarding his manufacturing facilities. A separate application needs to be submitted to each manufacturing site.
Application has to be submitted online via NMRA’s web portal eNMRA which requires uploading of essential information and documents to enable a desk review and GMP Inspections.
The NMRA Act, No. 5 of 2015 (section 51) & regulations 26 to 30 of part II of medicines regulations pronounce the legal requirements.
After these procedures, an approved Local manufacturer can submit application for formulation approvals.
After obtaining the formulation approval and GMP approval for commercial manufacturing of the facility, local manufacturer can apply for the product registration. After that, local manufacturer can apply for the manufacturing license.
Stages for beginners;
Site Master Files are submissions to NMRA used to provide confidential, detailed information about facilities, processes, or articles used in the manufacturing, processing, packaging, and storing of Medicines, Medical Devices, Borderline products and cosmetics.
References:
Good manufacturing practice (GMP) is that part of quality assurance which ensures that products are consistently produced and controlled to the quality standards appropriate to their intended use and as required by the marketing authorization.
The National Medicines Regulatory Authority Act No. 5 of 2015 interprets GMP guidelines as ‘good manufacturing guidelines issued by the World Health Organization’. As such, the National Medicines Regulatory Authority (NMRA) adopts the WHO GMP guidelines along with its subsequent updates for the purpose of regulation of pharmaceutical manufacture. The manufacturers are expected to adhere to the Good Manufacturing Practices throughout their activities.
Pharmaceutical manufacturers who intend to apply for marketing authorization in Sri Lanka may be subjected to one or several GMP inspections prior to their site approvals.
GMP Guidelines available on WHO website:
WHO good manufacturing practices for pharmaceutical products: main principles:
https://www.who.int/medicines/areas/quality_safety/quality_assurance/TRS986annex2.pdf?ua=1
WHO good manufacturing practices for sterile pharmaceutical products:
WHO good manufacturing practices for biological products:
https://www.who.int/publications/m/item/annex-2-WHO-gmp-for-biological-products
Guideline on Approval of an Overseas Manufacturing Plant of Medicines
Guideline on Preparation of Site Master Files
Guideline for preparation and conduct of the GMP inspection
Those who are willing to apply for Waiver of registration for Medical Device please be kind enough to submit your properly filled application form and relevant documents to office of Sate Ministry of Production, Supply and Regulations & Pharmaceuticals.

Download WOR Application from here : WOR Application
Documents Required for Waiver of Registration.
Mandatory Documents
Where Applicable
Note: Incomplete application will not be tabled to WOR Committee.

Healthcare professional such as doctors, dentists, pharmacists and nurses are encouraged to report suspected adverse events encountered in their day to day practice. Using the ADR google form given by the following link here, you can report Adverse Events.
Report Adverse Events
Address :
Director General / CEO
National Medicines Regulatory Authority,
No: 120, Norris Canal Road,
Colombo 10.
Telephone :
+94 112 698 896, +94 112 698 897
Fax :
+94 112 689 704
A copy of the form shall be forwarded to Adverse Drug Reaction Monitoring Unit of the Department of Pharmacology, Faculty of Medicine, University of Colombo, Kynsey Road, Colombo 08. (TP/ Fax: +94 112 697 483).
The Advertising Evaluation Sub Committee of the NMRA regulates advertisements - printed and electronic material - related to medicines, devices, borderline products to ensure such advertisements are ethical, accurate and appropriate.
Advertisement includes any representation by any means whatsoever, for the purpose of promoting directly or indirectly the manufacture, sale or disposal of any medicine medical device, borderline product.
All the applicants who which to obtain approval for the advertisement on medicines, medical device and borderline products should be followed below procedure.
1. Submit the duly filled application (Please mention email address correctly for sending evaluation sheet) along with copy of valid registration certificate, story board of the advertisement to the accepting counter on every Friday (From 9.00 am to 3.30 pm)for obtaining the payment note for processing fee (1000USD).
2. Payment Receipt issued by account division should be submitted NMRA after the payment along with the application with following documents to accepting pharmacist (Every Friday from 9.00 am to 3.30 pm). Application should be submitted in a file. Accepting pharmacist accepts the application and assign a reference number.
a. Duly filled and signed application
b. Copy of the valid registration certificates
c. 3 sets of printed copies/story board of the advertisement (If advertisement is containing any sound/ video, story board and 2 CD s including the video clip or sound clip need to be submitted)
d. Supportive documents (Clinical data, test report etc.) for proving any claims
3. All the applications will be tabled to the committee (Once a month) and evaluation reports will be emailed to the applicants.
4. Approved letters can be collected from the issuing counter.in the advertisements
Applicants should consider the following when designing an advertisement:
Medical Devices Pricing

101st MDEC Committee- (11th of February 2026)
MDEC Sub Committee- (21st of January 2026)
100th MDEC Committee- 07th of January 2026
99th MDEC Committee- (10th of December 2025)
MDEC Sub Committee- (21st of November 2025)
98th MDEC Committee- (06th of November 2025)
MDEC Sub Committee- (17th of October 2025)
97th MDEC Committee (01st of October 2025)
MDEC Sub Committee- (22nd of September 2025)
96th MDEC Committee (03rd of September 2025)
MDEC Sub Committee- (21st of August 2025)
95th MDEC Committee (06th of August 2025)
MDEC Sub Committee- (25th of July 2025)
94th MDEC Committee (09th of July 2025)
MDEC Sub Committee- (20th of June 2025)
93rd MDEC Committee (04th of June 2025)
92nd MDEC Committee (08th May 2025)
91st MDEC Committee (02nd of April 2025)
90th MDEC Committee (05th March 2025)
89th MDEC Committee (05th of February 2025)
88th MDEC Committee (08th of January 2025)
MDEC Sub Committee (24th of December 2024)
87th MDEC Committee (04th of December 2024)
MDEC Sub Committee (21th of November 2024)
86th MDEC Committee (06th of November 2024)
MDEC Sub Committee (24th of October 2024)
85th MDEC Committee (09th of October 2024)
MDEC Sub Committee (26th of September 2024)
84rd MDEC Committee - ( 04th of September 2024 )
MDEC Sub Committee - ( 22nd August 2024 )
83rd MDEC Committee - ( 07th of August 2024 )
82nd MDEC Committee (17th of July 2024 )
MDEC Sub Committee - ( 26th of June 2024 )
From MDEC - ( 10th of March 2022 to MDEC 5th of June 2022 )
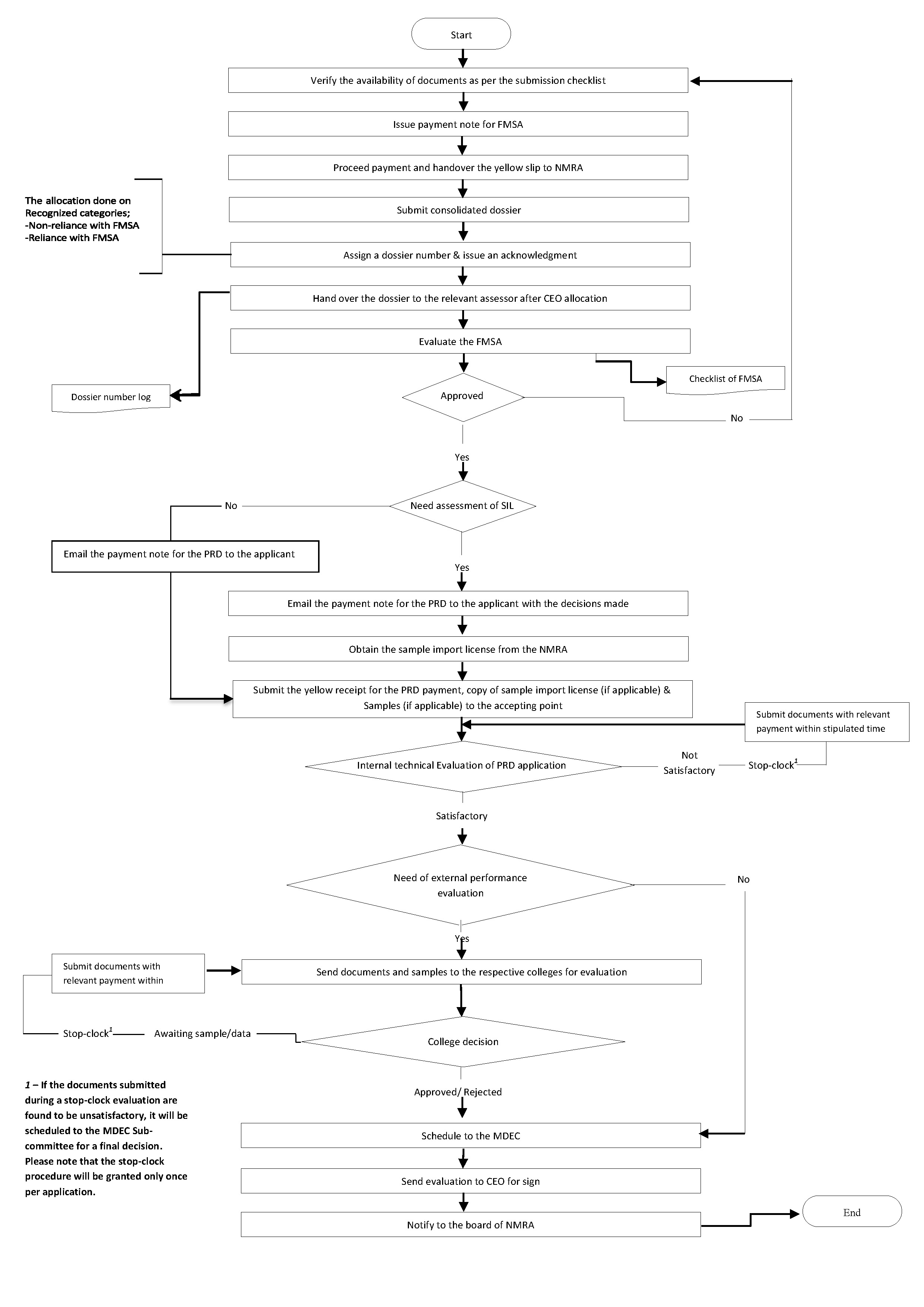
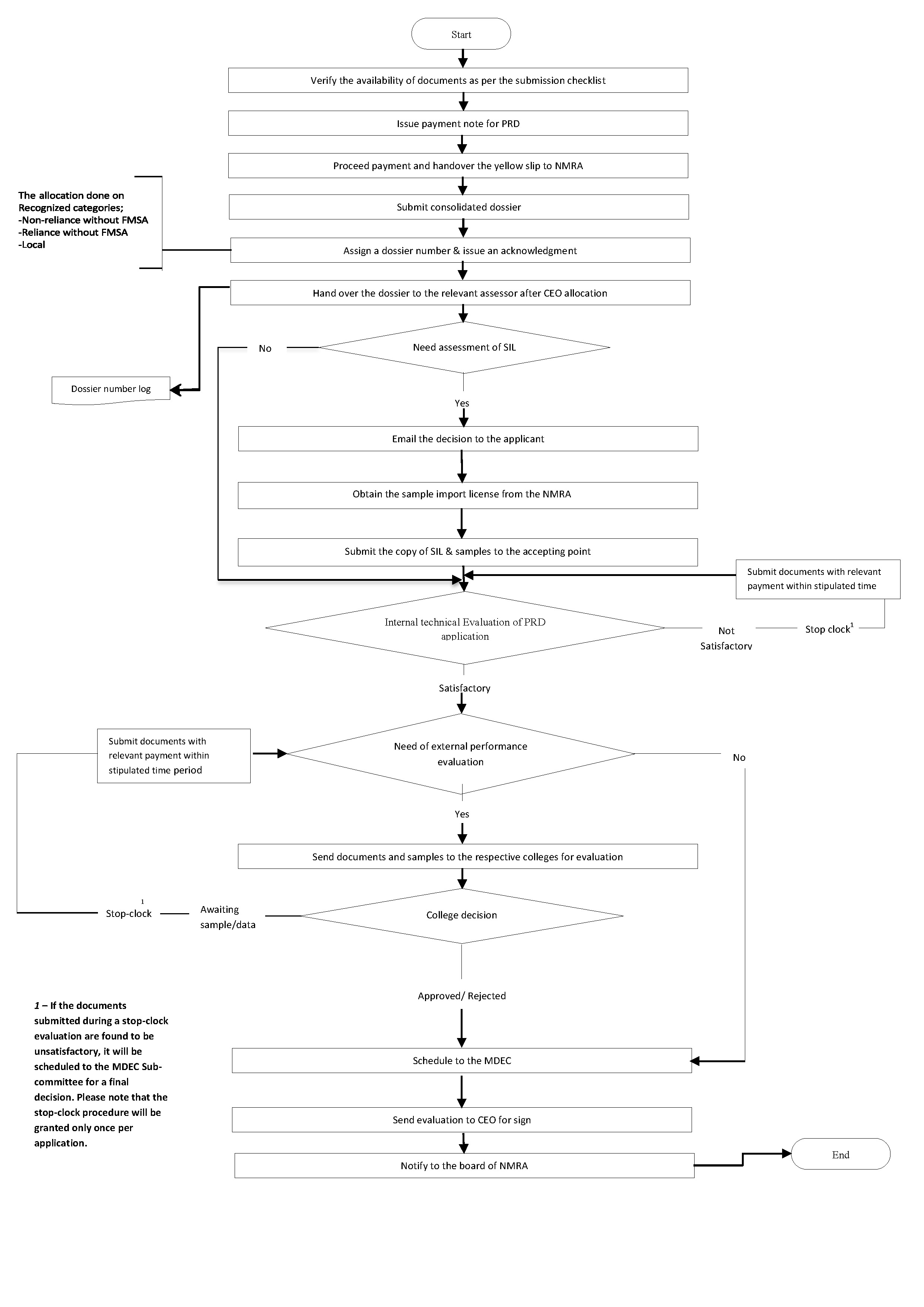
Procedure for submission of Dossier & other applications for Medical device registration
All dossiers and other applications for medical device registration will be accepted after applying by filling out the Google form linked below or scanning the provided QR code.

Fill out the following relevant checklist and submit two copies along with the dossier to the accepting point.
National Medicines Regulatory Authority,
State Engineering Corporation Building (2nd Floor),
No. 130,
W.A.D Ramanayaka Mawatha,
Colombo 02,
Sri Lanka